
【皓元·行研】当稳则稳,当断则断——更稳定的酶可裂解型Linker
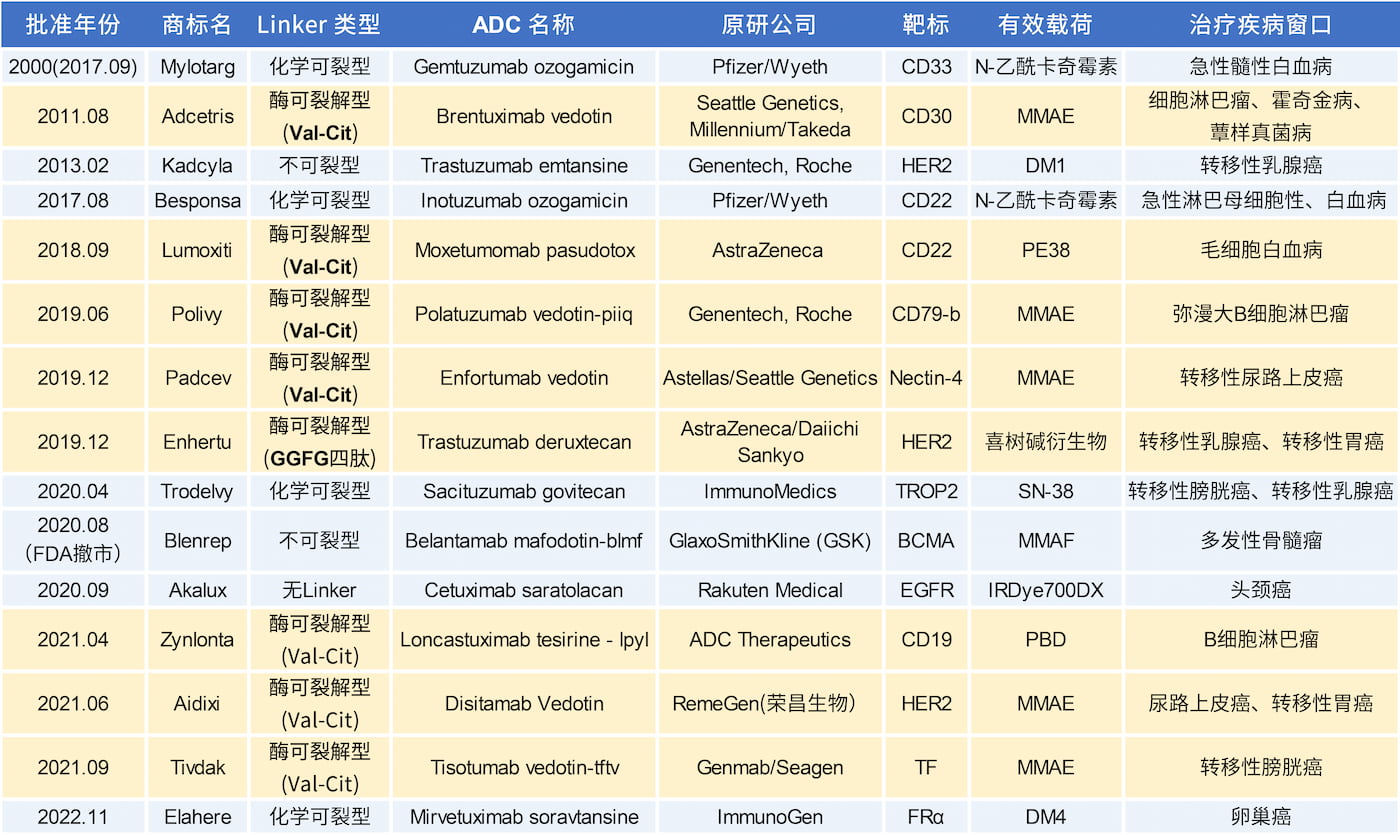
组成抗体偶联药物(ADC)的Linker片段一般分为可裂解和不可裂解两种类型[1]。最初,第一款 ADC药物 Mylotarg(麦罗塔)采用的是可裂解型Linker,由卡奇霉素通过一个包含腙键的酸可裂解Linker与Gemtuzumab偶联而成,于2000年被FDA批准用于治疗急性髓系白血病(AML)。腙键在弱碱性pH条件下理论上应保持稳定并且会选择性地在肿瘤相关的酸性环境下裂解。然而其在实际应用中却并不稳定,在血液循环中也会有部分发生断裂导致卡奇霉素过早释放,严重的肝毒性使得Mylotarg在2010年被退市。
为了克服Linker在体循环中的不稳定性,后来一些上市的ADC药物采用了不可裂解型Linker(如:Kadcyla 和 Blenrep)。不可裂解型Linker由稳定的化学键组成,在体循环中具有极好的稳定性,但是其在克服肿瘤异质性上有很大的局限性。
因为不可裂解型Linker对应ADC的药效发挥完全依赖于内吞机制,经肿瘤细胞内吞后ADC的抗体部分被降解,释放出包含毒素、Linker和氨基酸残基的复合物。然而带电荷的氨基酸复合物往往缺乏细胞渗透性,无法顺利穿过靶细胞膜的疏水性脂质双层来发挥旁观者效应[2]。缺少了旁观者效应的加持,靶抗原阳性细胞周围低表达或阴性的癌细胞将难以得到有效的杀伤[3],使得治疗始终受限于肿瘤的异质性。因此该类型Linker的ADC适用范围相对有限,目前主要用于对高抗原表达肿瘤或血液肿瘤的治疗[4]。
理想的Linker应在体循环中保持足够的稳定性,避免过早裂解释放毒素导致系统毒性,且能在目标位点高效地特异性裂解释放。当稳则稳,当断则断,看似矛盾的功能性要求正是Linker开发的主要挑战。
过去十年的研究重点是开发更稳定的酶可裂解型Linker[5]。据统计目前已上市的15款ADC药物中,有12款采用的是可裂解的Linker,其中8款是酶可裂解型Linker(如下图)。细胞溶酶体中存在的高浓度特异性水解酶为酶可裂解型Linker提供了绝佳的专一性裂解基础[2]。
组织蛋白酶B可裂解型Linker
组织蛋白酶B为半胱氨酸蛋白酶,通常与肿瘤进展有关在多种癌症中都观察到其过度的表达[2]。组织蛋白酶B是目前酶解机制中研究和临床开发最多的,同时也是目前唯一有实际应用于上市ADC药物中的Linker裂解酶,其主要针对肽连接子(如:Val-Cit、Val-Ala和GGFG等)。含有Val-Cit的Linker是上市ADC中应用最多的二肽连接子,如:Adcetris、Polivy、Padcev、Tivdak、Aidixi、Lumoxiti和Zynlonta采用的都是含Val-Cit的Linker。Val-Ala已成为一种有效的可裂解基团,在独立的组织蛋白酶B裂解实验中Val-Ala像Val-Cit一样在细胞中可以有效地裂解,并且在人血浆中高度稳定。虽然Val-Ala的裂解速率是Val-Cit的一半,但其疏水性较低,这一点的重要性在ADC的偶联中得到了体现。在Val-Ala的案例中,每个mAb平均装载了7.4个药物分子且并没有发生显著的聚集现象,而含Val-Cit的Linker由于聚集和沉淀过多,不能达到DAR > 4[6]。

其它早研阶段的酶可裂解型Linker
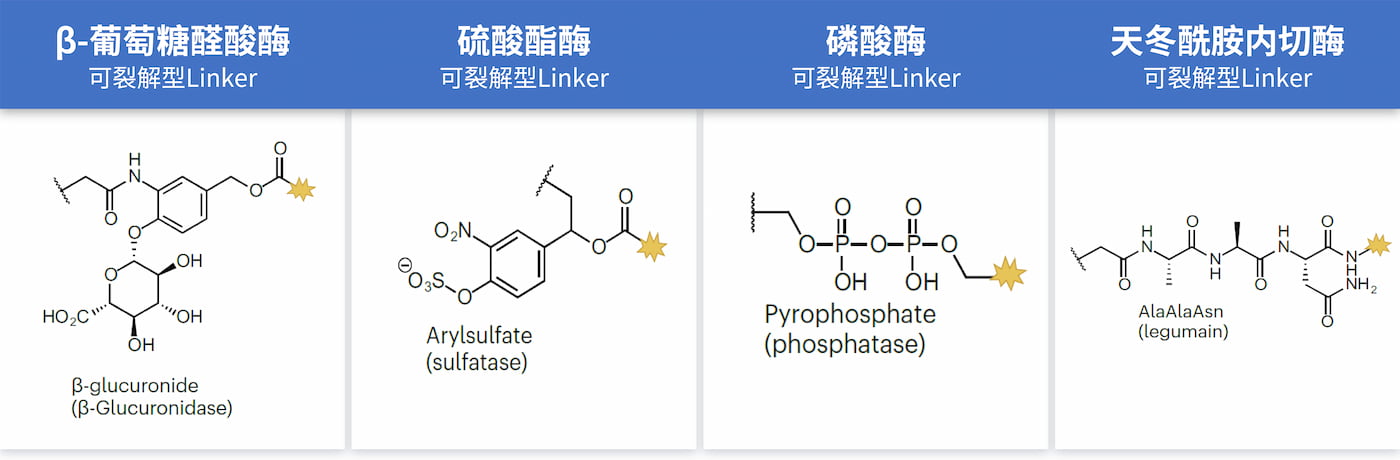
▲ 图片来源于参考文献[5]
其他处于早期研究阶段的还有:β-葡萄糖醛酸酶[7,8,9]、硫酸酯酶[10]、磷酸酶[11]和天冬酰胺内切酶[12,13]可裂解型Linker等(如图)。β-葡萄糖醛酸酶和磷酸酶可裂解的Linker已经显示出良好的溶解性和稳定性[2],因此其在ADC的未来临床前景中有望进一步发挥作用。同时,随着更深入的发展,人们发现越来越多的与肿瘤微环境相关的酶也可被用来实现此类Linker在肿瘤细胞外区域的裂解释放。由于此类Linker具有在肿瘤细胞内外都可释放的优势潜力,使得相应抗原的可选范围得到了进一步的扩大[2]。
总结
由于旁观者效应对于进一步提高对肿瘤细胞杀伤力的重要性,可裂解型Linker是治疗大多数肿瘤的首选。酸依赖的化学可裂解型Linker起初很有潜力,但是随着高效细胞毒素的开发对体循环稳定性更高的要求,它们的实际的应用价值被降低。相比之下,酶可裂解型Linker能够更有效地区分血浆和靶细胞条件,并且正在进一步发展以用于细胞外裂解的作用模式。通过消除对抗原内化的需求,这种替代作用机制大大增加了可能的抗原靶点数量,这为更多新的ADC疗法打开了大门。随着未来对Linker旁观者效应的研究以及细胞外裂解作用机制的探索,这类酶可裂解型Linker的整合式创新有望更进一步地持续驱动ADC的升级。
参考文献:
[1] Z. Su, et al., Antibody-drug conjugates: Recent advances in linker chemistry, Acta Pharm Sin B, 2021, 11(12), 3889-3907.
[2] J. D. Bargh, et al., Cleavable linkers in antibody–drug conjugates, Chem Soc Rev., 2019, 48, 4361-4374.
[3] Y. V. Kovtun, et al., Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen, Cancer Res., 2006, 66, 3214–3221.
[4] R. V. J. Chari, et al., Antibody–Drug Conjugates: An Emerging Concept in Cancer Therapy, Angew. Chem., Int. Ed., 2014, 53, 3796–3827.
[5] K. Tsuchikama, et al., Exploring the next generation of antibody-drug conjugates, Nat. Rev. Clin. Oncol., 2024, 21, 203–223.
[6] S. C. Jeffrey, et al., Dipeptide-based highly potent doxorubicin antibody conjugates, Bioorg. Med. Chem. Lett., 2006, 16, 358–362.
[7] P. J. Burke, et al., Glucuronide-linked antibody–tubulysin conjugates display activity in MDR+ and heterogeneous tumor models, Mol. Cancer Ther. 17, 1752–1760 (2018).
[8] S. C. Jefrey, et al., Development and properties of beta-glucuronide linkers for monoclonal antibody–drug conjugates, Bioconjug. Chem. 17, 831–840 (2006).
[9] S. Chuprakov, et al., Tandem-cleavage linkers improve the in vivo stability and tolerability of antibody–drug conjugates, Bioconjug. Chem. 32, 746–754 (2021).
[10] J. D. Bargh, et al., Sulfatase-cleavable linkers for antibody–drug conjugates, Chem. Sci. 11, 2375–2380 (2020).
[11] J. C. Kern, et al., Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates, J. Am. Chem. Soc. 138, 1430–1445 (2016).
[12] H.G. Lerchen, et al., Tailored linker chemistries for the eficient and selective activation of ADCs with KSPi payloads, Bioconjug. Chem. 31, 1893–1898 (2020).
[13] J. T. Miller, et al., Enzyme-agnostic lysosomal screen identifies new legumain-cleavable ADC linkers, Bioconjug. Chem. 32, 842–858 (2021).